ChIP partitioning using different datasets
Part I: Simulated data [To be performed in R]
Composite peak data
K=2; L=63; r=list(1:21,22:42,43:63) C=matrix(nrow=K, ncol=sum(L)) C[1,r[[1]]] = 2*dnorm(1:21, 8,1.0) C[1,r[[2]]] = 5*dnorm(1:21, 8,3.0) C[1,r[[3]]] = 1*dnorm(1:21,11,0.5) C[2,r[[1]]] = 4*dnorm(1:21, 8,3.0) C[2,r[[2]]] = 3*dnorm(1:21,14,1.0) C[2,r[[3]]] = 1*dnorm(1:21, 8,0.5) Q = c(0.6,0.4)Definition of random samples
N=10000
samples=matrix(data=0, nrow=N, ncol=K)
rs=sample(1:K,N,rep=T,prob=Q)
for(i in 1:N) {samples[i,rs[i]]=1}
Random data generation
data=matrix(nrow=N, ncol=L)
for (i in 1:N) {data[i,] = rpois(L,C[which(samples[i,] == 1),])}
# Let's have a look at the aggregation plot of the complete set
plot(colMeans(data), type = "l")
Load the em_functions.R file:
source("em_functions.R")
Partioning with random seeds
c=matrix(nrow=L, ncol=K)
p=matrix(nrow=N, ncol=K)
for(i in 1:K) {p[,i] = rbeta(N,N**-0.5,1)}
c = (t(p) %*% data)/colSums(p)
q=rep(1/K,K)
for(i in 1:20) {em_shape(c,q,data); plot_classes(c); print(q)}
Iterative partitioning
c = matrix(data=colMeans(data), nrow=1, ncol=L)
flat =matrix(data= mean(data), nrow=1, ncol=L)
q = 1
for (m in 1:(K-1)) {
c = rbind(flat,c)
q = c(1/m,q); q = q/sum(q)
for(i in 1:20) {em_shape(c,q,data); plot_classes(c); print(q)}
}
Iterative partitioning with untrained flat class
c = matrix(data=colMeans(data), nrow=1, ncol=L)
flat =matrix(data= mean(data), nrow=1, ncol=L)
q = 1
for (m in 1:(K)) {
c = rbind(flat,c)
q = c(1/m,q); q = q/sum(q)
for(i in 1:20) {
c[1,]=flat; em_shape(c,q,data); plot_classes(c); print(q)}
}
cor(p, samples) # Pearson's correlation between the real and predicted class probabilities
Part II: CTCF dataset [GEO series GSE29611 Histone Modifications by ChIP-seq from ENCODE/Broad Institute]
- ENCODE peak list for CTCF from HUVEC cell line. Download the peak list.
- Process tags data from HUVEC cell line. We will use it directly from the server.
- Reference feature: above peak list
- Target feature: hg19; ENCODE ChIP-seq; GSE29611; HUVEC CTCF (do appropriate centring)
- Analysis parameters: -500 to 500; Window width:10; Count cut-off:1
- bed to sga for p-value classes
- OProf Parameters: -499 to 500; Window size: 50, Window shift: 1; Search mode: bidirectional; Cut-off: p-val: 1.0000e-05
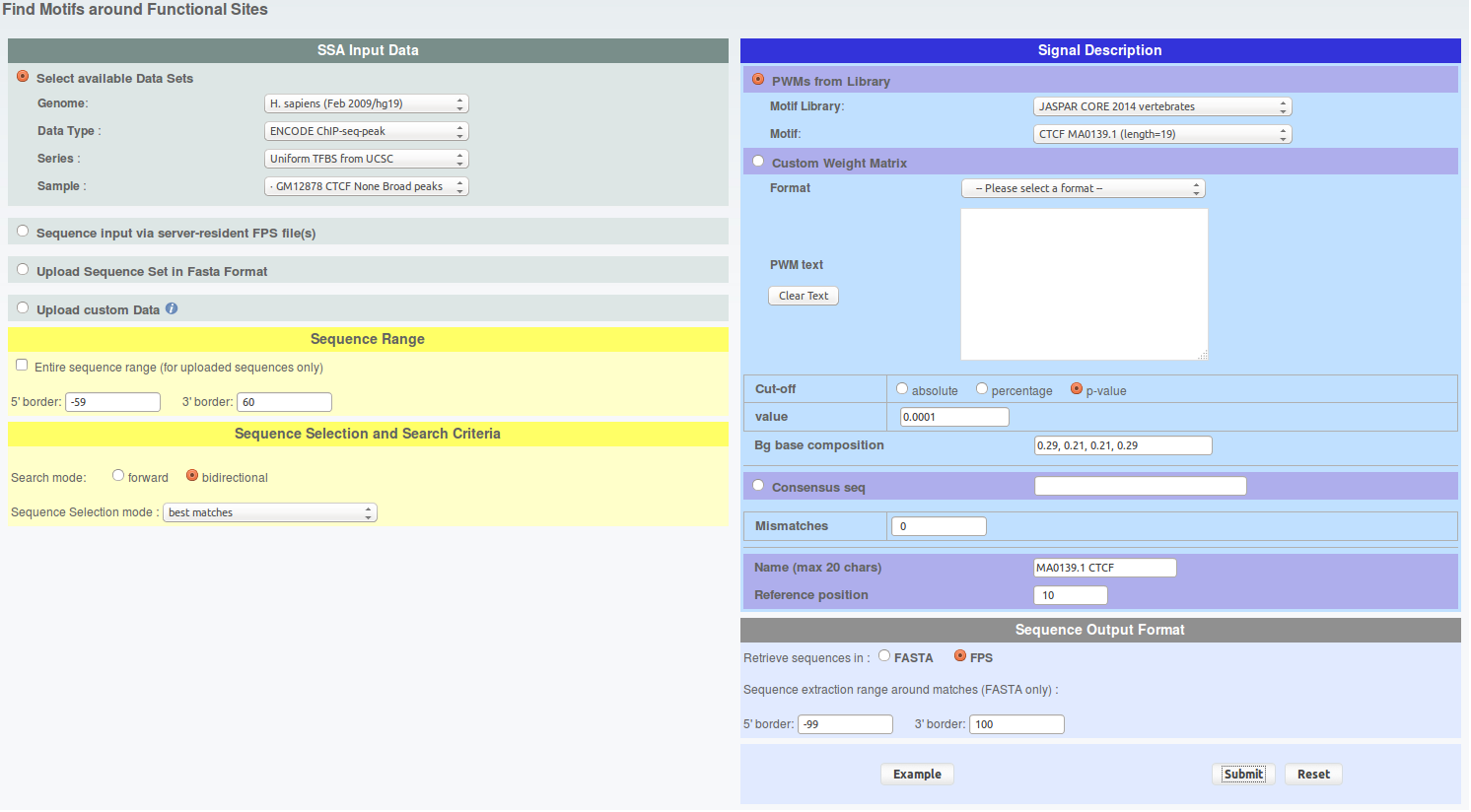
- PWM from library: JASPAR CORE 2014 vertebrates; Motif: CTCF MA0139.1 (length=19)
In the current exercise we will be trying to reproduce one of the figures from following paper:
Nair et al., 2014, Probabilistic partitioning methods to find significant patterns in ChIP-Seq data, Bioinformatics, 30, 2406-2013, PMID 24812341
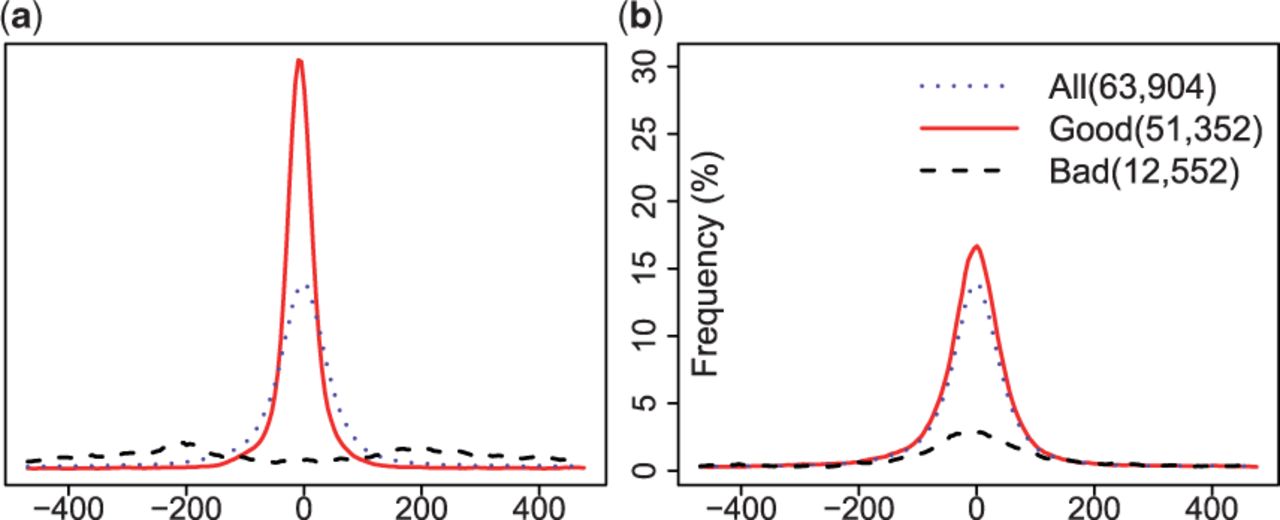
Shape-based peak evaluation with shifting. The figure illustrates the effects of probabilistic partitioning on a CTCF peak list provided by ENCODE in terms of motif enrichment. (a) Probabilistic partitioning with shifting. (b) Partitioning based on original P-values.
In order to reproduce the above figure, we will need an ENCODE peak list and associated tags data.
Pre-processing of data to generate tag count matrix.
We will use ChIP-Extract Analysis Module to generate a tag count matrix in defined bins around CTCF sites. Select appropriate target feature and then click submit.
Download the Ref SGA File and Table (TEXT) and save as huvec_peaks.sga and huvec_ctcf.txt respectively.
Read data:
data = as.matrix(read.table("huvec_ctcf.txt"))
Define classes and shifts:
K=1; S=11; N=dim(data)[1]; L=dim(data)[2]-S+1; ITER=10Iterative shape based partitioning with shifting
c = colMeans(data[,(floor(S/2)+1):(floor(S/2)+L)])
flat = matrix(data=mean(data), nrow=1, ncol=L)
q=q0 = dnorm(1:S,floor(S/2)+1,1)/sum(dnorm(1:S,floor(S/2)+1,1))
for (m in 1:(K)) {
c = rbind(flat,c)
q = rbind(q0/m,q); q/sum(q)
for(i in 1:ITER) {reg_shift(q); c[1,]=flat;
reg_shift(q); em_shape_shift(c,q,data); plot_classes(c); print(q)}
}
prob = rowSums(p[,2,])
shift = apply(p,c(1,3),sum) %*% (1:S)
Read peaks and filter based on probability value into different classes:
peak <- read.table("huvec_peaks.sga", header=F, sep="\t")
good <- peak[which(prob>=0.5),]
bad <- peak[which(prob<=0.5),]
peak[,3] <- round(peak[,3]+10*(shift-6))
good_shifted <- peak[which(prob>=0.5),] # good peaks after shifting
Write output as sga files.
write.table(good, "chipPart_good.sga", sep="\t", quote=F, row.names=F, col.names=F) write.table(bad, "chipPart_bad.sga", sep="\t", quote=F, row.names=F, col.names=F) write.table(good_shifted, "chipPart_good_shifted.sga", sep="\t", quote=F, row.names=F, col.names=F)Read the bed file and sort based on p-value generated by ENCODE consortium. For fair comparison, the threshold for partitioning with the original p-values is chosen such as to match the numbers of good and bad peak obtained with probabilistic partitioning.
pval <- read.table("ctcf_peaks.bed", header=F, sep="\t")
spval <- pval[order(pval[,8], decreasing=T),]
goodpval <- spval[1:nrow(good),]
badpval <- spval[nrow(good)+1:nrow(pval),]
write.table(goodpval, "pval_good.bed", sep="\t", quote=F, row.names=F, col.names=F)
write.table(badpval, "pval_bad.bed", sep="\t", quote=F, row.names=F, col.names=F)
Use ChIP-Convert to convert the output of partitioning into following formats.
Following piece of experiment needs to be carried out on SSA web-server. Extract CTCF motif enrichment (for all the sga files produced in previous step) using Oprof
Save the graph output as: good_part.txt, bad_part.txt, good_shifted_part.txt, high_pvalue.txt, low_pvalue.txt and ctcf_all.txt (for all the sites)
Now move back into R for plotting the results. This will produce the figure as shown above.
## read files that were the output/ graph text output from Oprof and plot results:
a <- read.table("good_part.txt", header=T, sep="")
b <- read.table("bad_part.txt", header=T, sep="")
c <- read.table("good_shifted_part.txt", header=T, sep="")
d <- read.table("high_pvalue.txt", header=T, sep="")
e <- read.table("low_pvalue.txt", header=T, sep="")
all <- read.table("ctcf_all.txt", header=T, sep="")
par(mar=c(0,2.75,1,1), mfrow = c(1,2), oma=c(4,0,2,0))
plot(c[,1],c[,2],type="l",lwd=2, col="red", cex.axis=1, xlab="", ylab="", yaxt="n", ylim=c(0,30));
points(c[,1],all[,2],type="l",lwd=2, col="blue", lty=4)
points(c[,1],b[,2],type="l",lwd=2, lty=3)
plot(d[,1],d[,2],type="l",lwd=2, col="red", cex.axis=1, xlab="", ylab="", ylim=c(0,30));
title(ylab="Frequency (%)", cex.lab=1, mgp=c(-2.2,2,1))
points(c[,1],all[,2],type="l",lwd=2, col="blue", lty=4)
points(c[,1],e[,2],type="l",lwd=2, lty=3)
legend("topright",c("All(63,904)","Good(51,136)","Bad(12,768)"),col=c("blue","red","black"), lwd=c(1,1,1), cex=1, box.col=NA, bty="n", lty=c(4,1,3), seg.len=2)
Part III: Nucleosome organization around transcription factor binding sites in GM12878 cells. For this exercise, the datasets come from the following sources : ENCODE Uniform, GSE32970, GSE35586, GSE29611
EPDHere are the needed R functions em_api.R. Download this file and store it in the directory you will work in. The code below contains calls to these functions.
In this exercice, we will investigate the nucleosome organization around CTCF and ETS1 binding sites in GM12878 cells. CTCF is a factor which is known to show nicely phased nucleosomes arrays
on each side of its binding sites. ETS1 is a transcription factor which is mostly found near transcription start sites (TSS) but which does not behave as CTCF regarding nucleosome organization.
We will first extract CTCF and ETS1 peaks from peak lists which have the corresponding factor motif in their neighbourhood using FindM. Then, we will measure the density of MNase and DNAseI reads
around these peaks by generating matrices of tag counts using ChIP-Extract. We will import these data in R and perform a probabilistic partitioning to try to identify several nucleosomes
organization classes around transcription factor binding sites and isolate classes of interest. Finally, we will extract one class of interest and go back to ChIP-Cor for additional analyses.
-
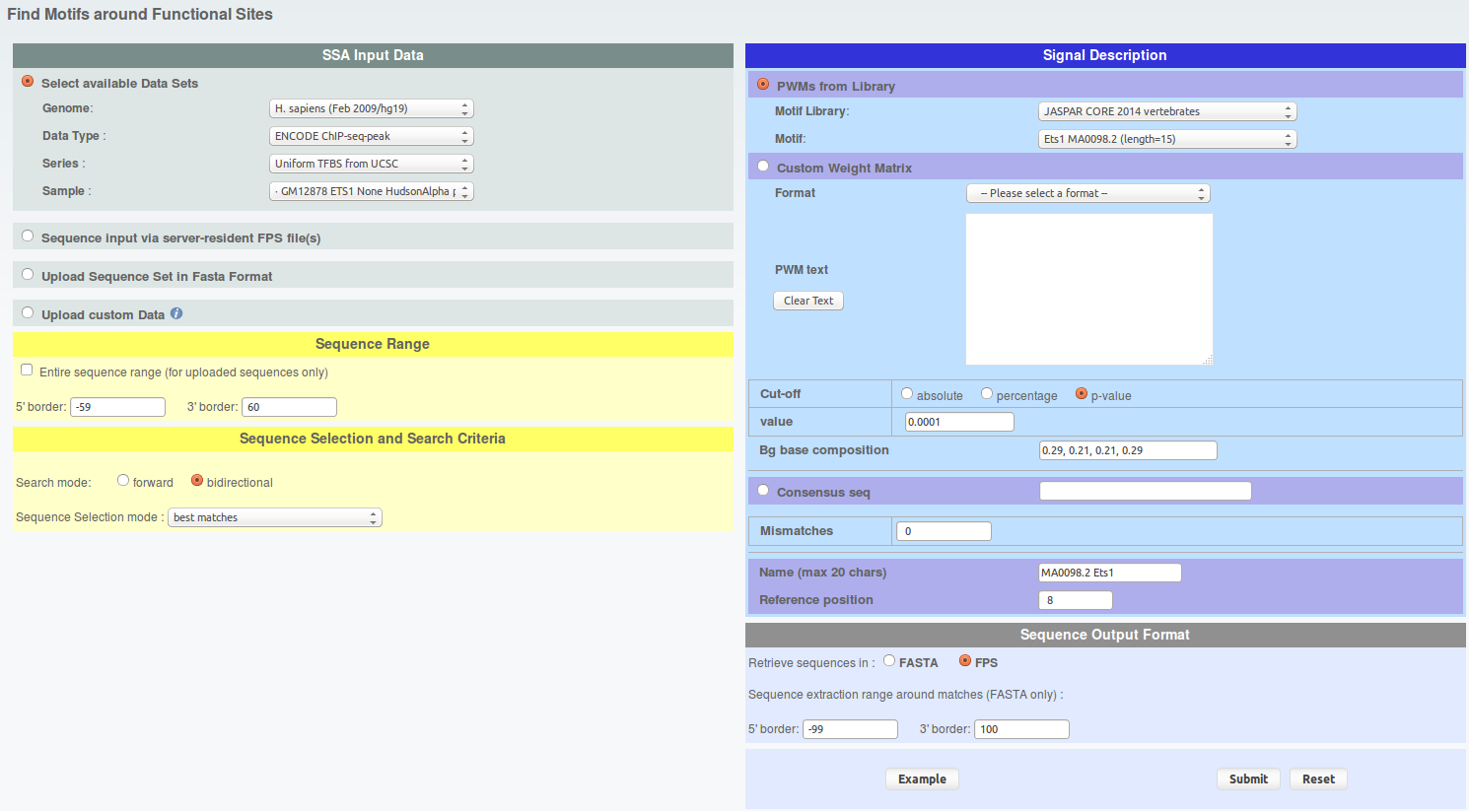
First, let's extract CTCF and ETS1 peaks which have CTCF and ETS1 binding motif nearby, respectively. To do this, go the FindM and fill the parameters as follows:
click to show/hide FindM parameters.From the result page you will need to do two things. First save the results on your hard drive under sga format and name the files 'findm_CTCF.sga' and 'findm_ETS1.sga' (in case you can retrieve these files here : findm_CTCF.sga findm_ETS1.sga). Second, redirect the results toward ChIP-Extract by clicking on the corresponding button under 'Useful links for Downstream Analysis'.
-
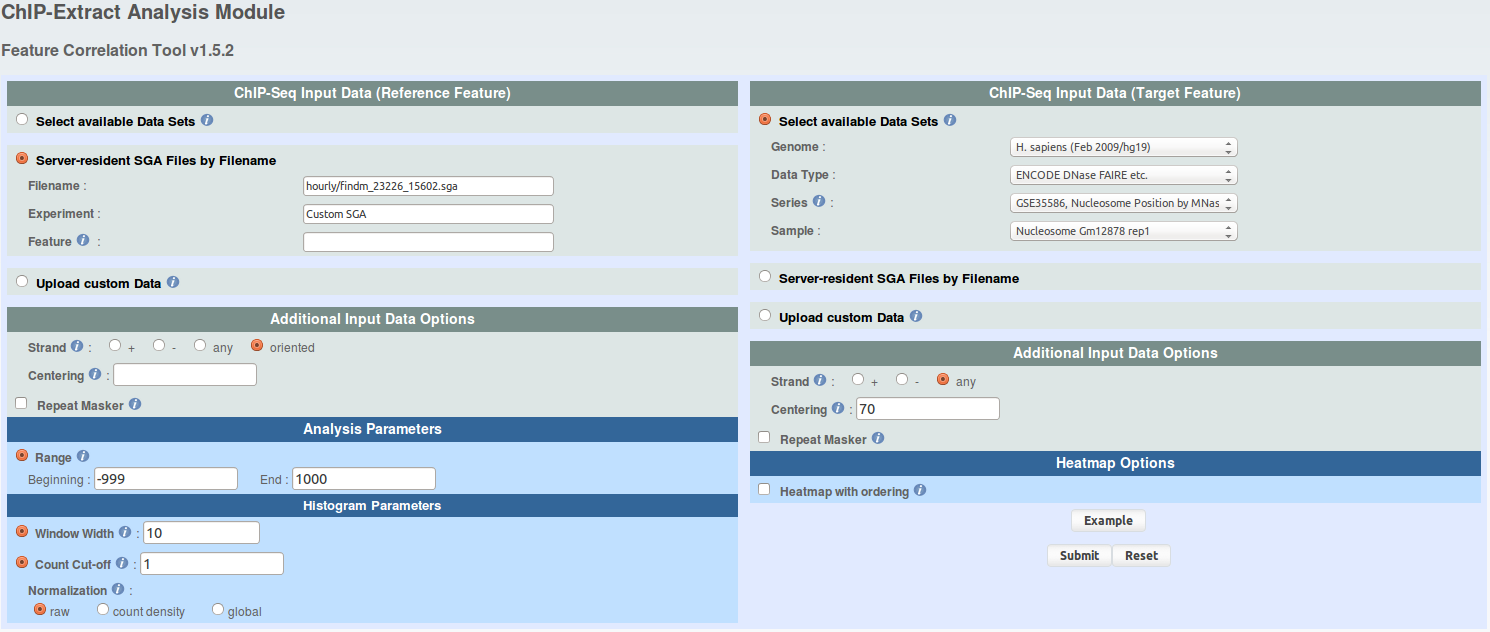
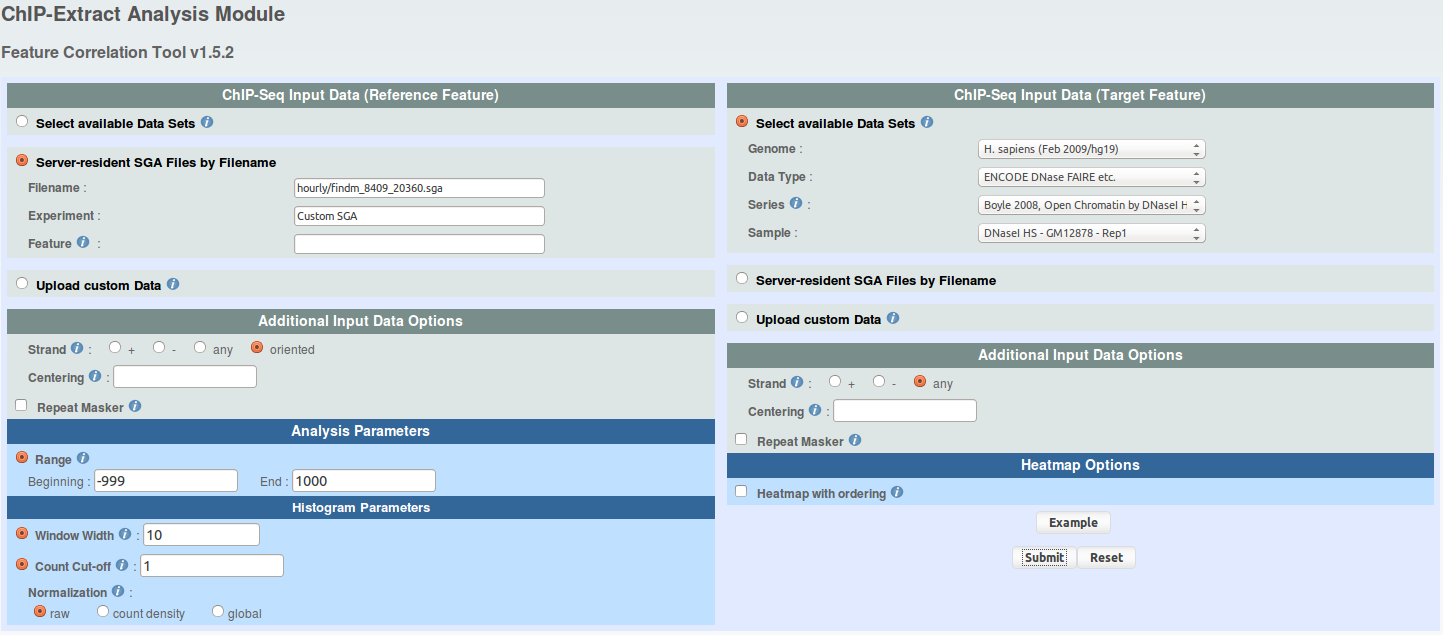
For each FindM result, run ChIP-extracts (4 runs total) as shown under and save ChIP-Extract results on your hard drive by naming your files 'chipextract_CTCF_MNase.mat','chipextract_CTCF_DNaseI.mat', 'chipextract_ETS1_MNase.mat', 'chipextract_ETS1_DNaseI.mat'.
click to show/hide ChIP-Extract parameters. -
Now open a R session in the folder where you have saved all the files and load the ChIP-Extract data.
click to show/hide R code. -
To verify whether CTCF has nicely phased nucleosomes arrays on each sides of its binding sites, and that ETS1 does not, you can draw aggregation plots. An aggregation plot is the type of plot that ChIP-cor returns you. ChIP-Extract data can be used to draw exactly the same plots as you would have had with ChIP-Cor.
click to show/hide R code. -
Now, let's run the EM partitioning to classify MNase signal and highlight whether different types of nucleosome organization are present in the data. We will allow the algorithm to shift and flip the data.
Nucleosome signal is perdiodic by nature. Allowing the vectors to be phased by shifting them will increase the resolution of the signal. For instance, if the nucleosomes are not always organized the same around the binding site, this can create interference and mess up the nucleosome signal. Hopefully, this can be fixed by allowing shifting. Additionally, allowing algorithm to flip the vector might help resolving symetrical patterns which may originate from the overlap of two asymetrical anti-sense patterns.
click to show/hide R code.click to show/hide the figures.
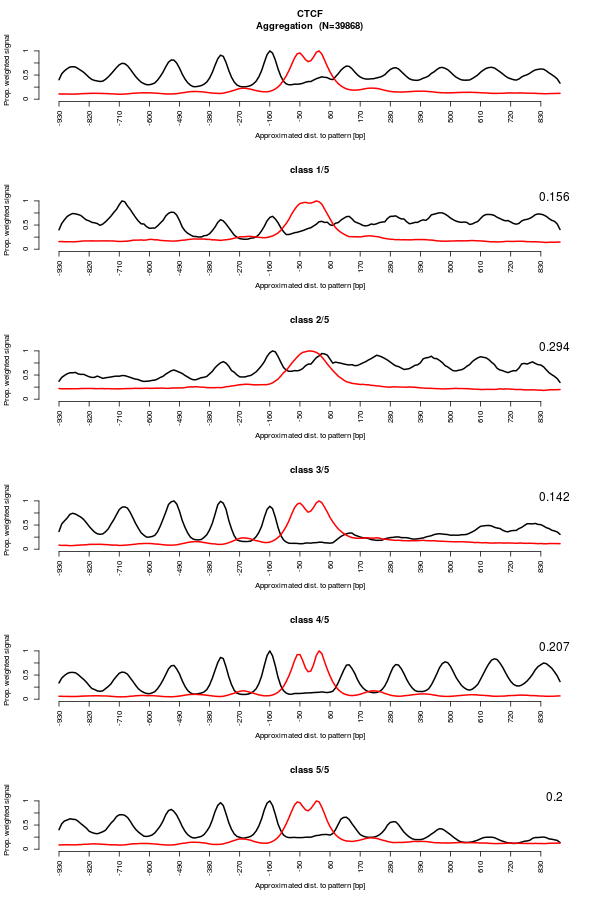
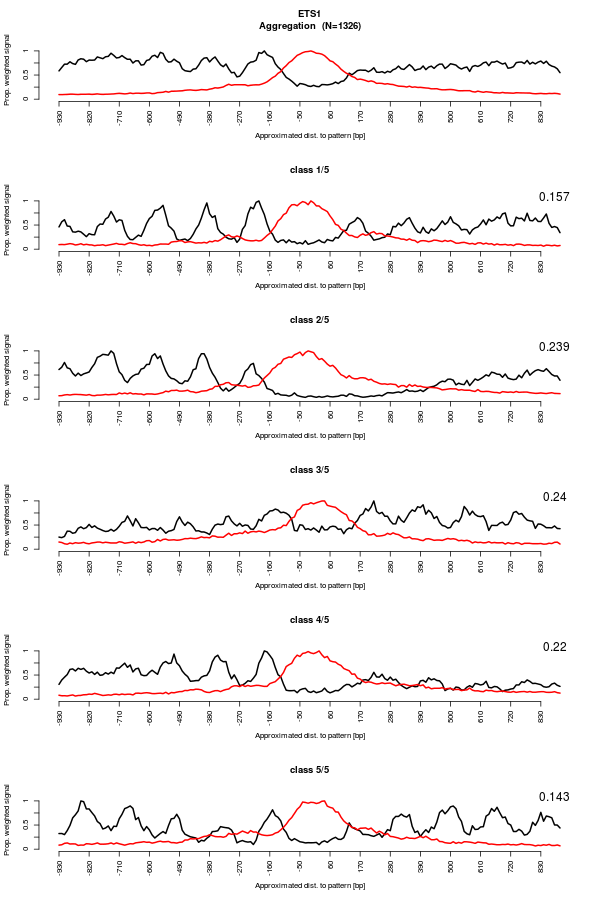
The result figures shows a few interesting things.
Regarding CTCF, a strong periodic MNase signal is still visible. Some classes are asymetric, suggesting that the CTCF sites may have an orientation. Moreover, the different classes show pretty different patterns, supporting different type of nucleosome organization around CTCF binding sites. Interestingly, the DNaseI footprint is still visible which indicates that the data were not shifted too much. This suggests that the nucleosomes are organized with respect to the binding sites (or a feature really close).
Regarding ETS1, we now see nucleosomes clearly visible. However, in contrast to CTCF, DNaseI footprint dissapeared and the DNaseI hypersentitivty region is now wider. This reflects the fact that the MNase data vectors have been shifted. This means that the first nucleosome is not a constant distance from the binding site. This suggests that the nucleosomes are not organized with respect to the binding sites. Additionally, unlike what the aggregation plot was showing, the nucleosome organization around ETS1 binding sites is not anymore symetrical. For instance, class 1 and 3 have a nucleosome array on one side of the binding site only. -
This algorithm performs a probabilistic (or soft) classification. This means that each binding site is assigned to all classes at the same time, with different probabilities. Nonetheless, it is still possible to extract all binding sites which have been assigned to a class, with a given minimal probability. Interestingly, it is also possible to update the original binding site position to take into account the shift and flip performed by the algorithm. In case, you can download the classification result file here : CTCF_5class_flip_15shift_random.rds ETS1_5class_flip_15shift_random.rds
We know that ETS1 is a factor that mostly bind near TSS. Class 1 shows a profile which is compatible with this. The nucleosome array might reflect the nucleosomes downstream the TSS with the commonly described nucleosome depleted region near the TSS. Let's extract the binding sites which have been classified has belonging to class 1 with an overall probability of 0.9 or higher.
click to show/hide R code. -
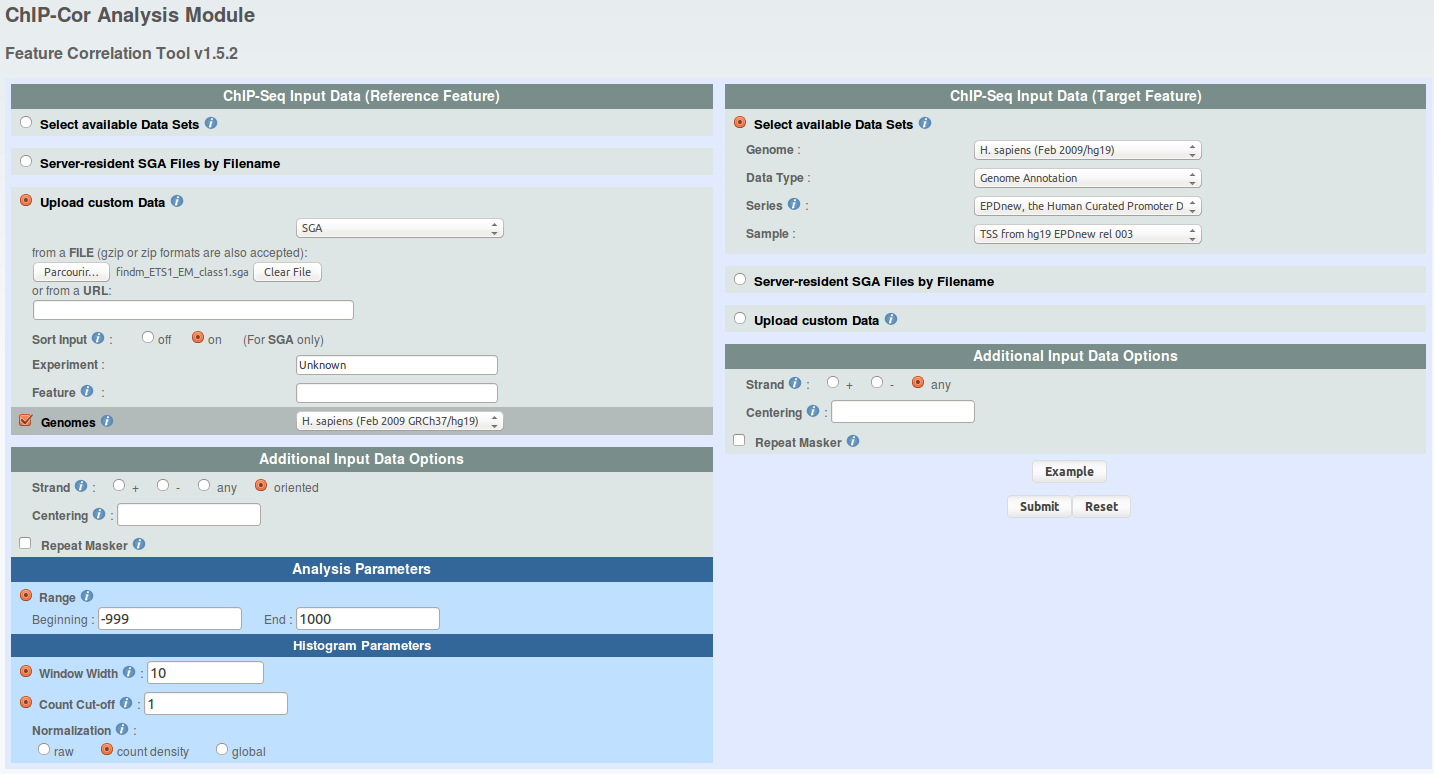
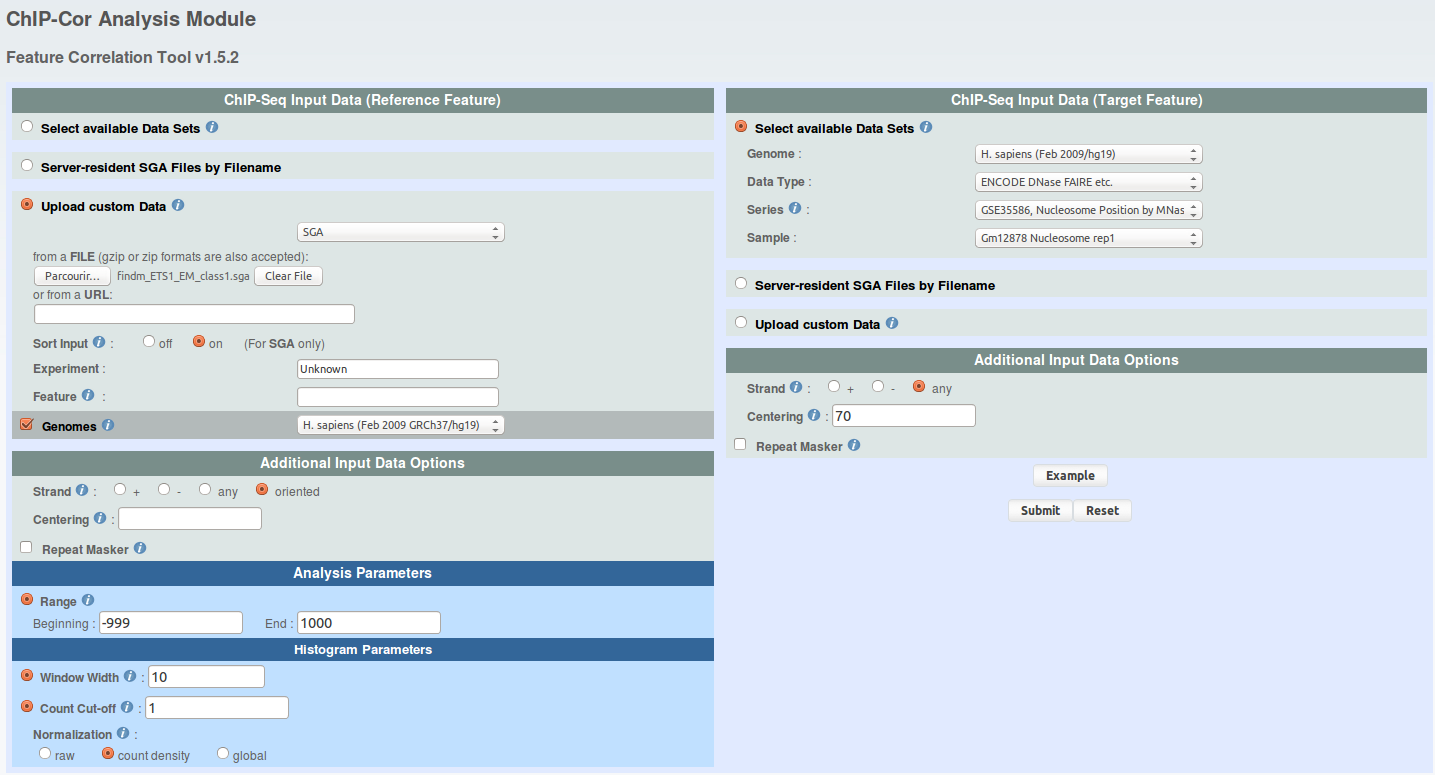
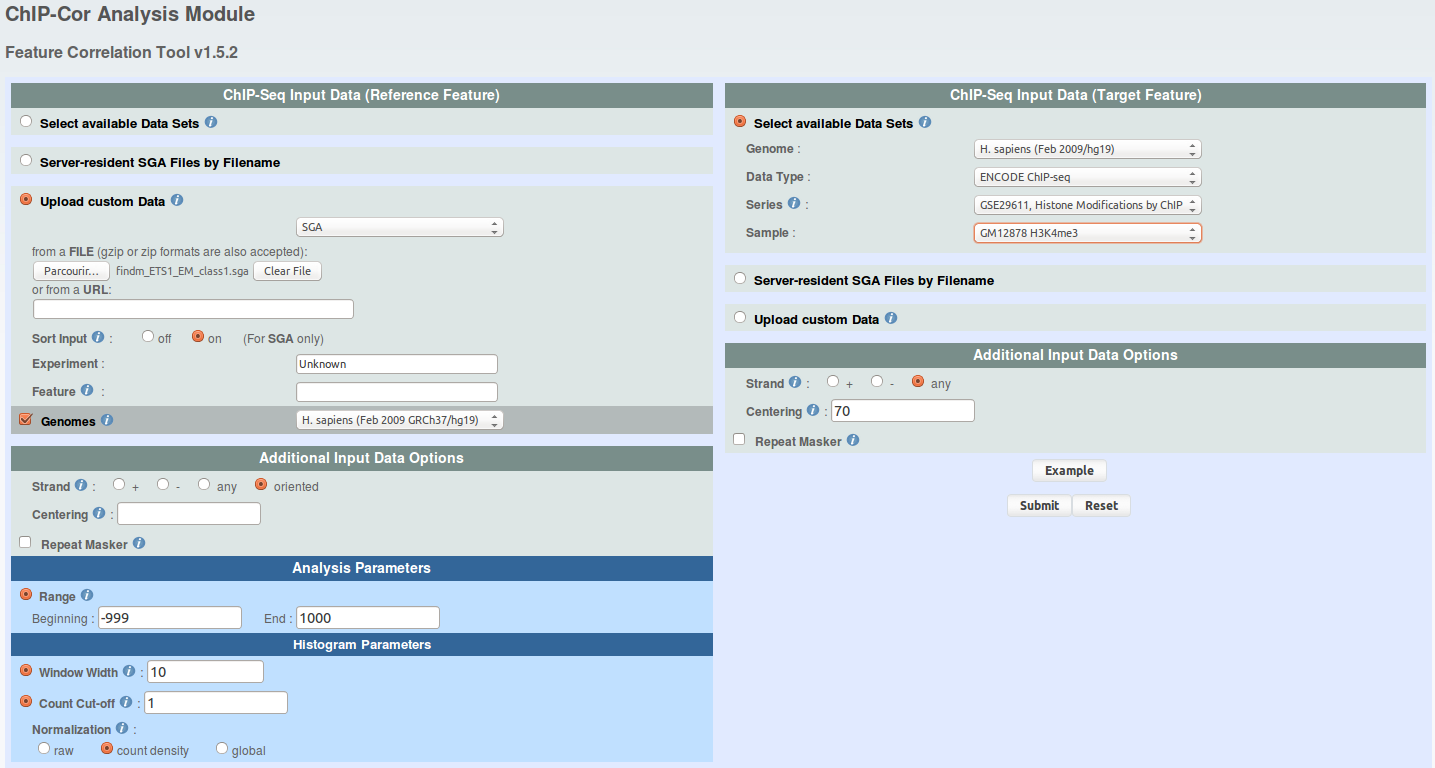
Now, go on ChIP-Cor, upload the coordinates extracted from class 1 and run ChIP-cor 3x as indicated under and save the results in the files named as follows : 'findm_ETS1_EM_class1_TSS.dat', 'findm_ETS1_EM_class1_MNase.dat', 'findm_ETS1_EM_class1_H3K4me3.dat'
click to show/hide ChIP-Cor parameters. -
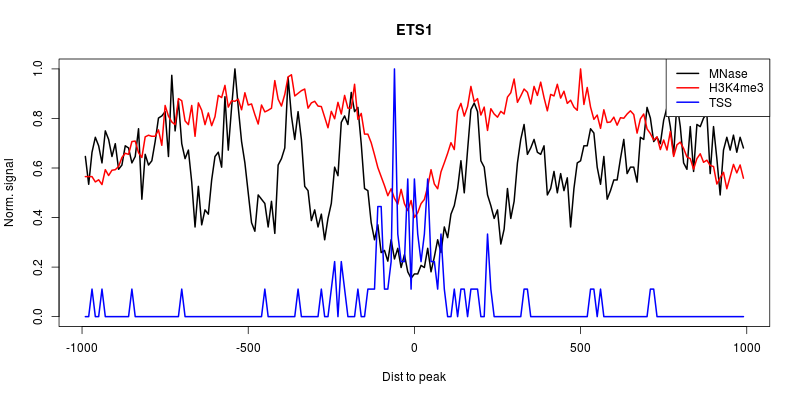
Go back to your R session and draw an aggregation plots with all the ChIP-Cor data.
click to show/hide R code.click to show/hide the figure.
The peak of TSS density in the nucleosome depleted region together with the H3K4me3 labelling suggest that the binding sites which have been assigned to this class are indeed in promoters regions. However, the exact orientation of these promoters is hard to estimate since H3K4me3 labelling is pretty symetrical and that we can see nucleosomes on both sides of the TSS density peak.
Now, you might want to pursue this analysis by extracting other classes coordinates or to rerun an entire analysis on other type of data such as histone marks.
Part IV: H3K4me3 around human promoters. Dataset in this example is from GEO series GSE29611 and EPD.
- Reference feature: hg19; Genome Annotation; EPDNew; rel 003
- Target feature: hg19; ENCODE ChIP-seq; GSE29611; GM12878 H3K4me3
- Analysis parameters: -1000 to 1000; Window width:10; Count cut-off:10
We will use ChIP-Extract Analysis Module to generate a tag count matrix in defined bins around promoters. Select appropriate target feature and then click submit.
Download the Ref SGA File and Table (TEXT) and save as promoters.sga and h3k4me3_promoters.txt respectively.
Read data:
data = as.matrix(read.table("h3k4me3_promoters.txt"))
Define classes and shifts:
K=4; S=3; N=dim(data)[1]; L=dim(data)[2]-S+1; ITER=5Iterative shape based partitioning with shifting
mean_shift=floor(S/2)+1
c = colMeans(data[,mean_shift:(mean_shift+L-1)])
flat = matrix(data=mean(data), nrow=1, ncol=L)
q=q0 = dnorm(1:S,mean_shift,1)/sum(dnorm(1:S,mean_shift,1))
for (m in 1:(K-1)) {
c = rbind(flat,c)
q = rbind(q0/m,q); q/sum(q)
for(i in 1:ITER) {
reg_shift(q); em_shape_shift(c,q,data); plot_classes(c); print(q)}
}
Extract class labels
class=apply(apply(p,c(1,2),sum),1,order)[4,]Write output sga files for different classes
peak <- read.table("promoters.sga", header=F, sep="\t")
write.table(peak[which(class==1),], "h3k4me3_class1.sga", sep="\t", quote=F, row.names=F, col.names=F)
write.table(peak[which(class==2),], "h3k4me3_class2.sga", sep="\t", quote=F, row.names=F, col.names=F)
write.table(peak[which(class==3),], "h3k4me3_class3.sga", sep="\t", quote=F, row.names=F, col.names=F)
write.table(peak[which(class==4),], "h3k4me3_class4.sga", sep="\t", quote=F, row.names=F, col.names=F)
Now you may want to explore these four classes in-terms of associated features, such as PolII, conservation, or check the enrichment for various core promoter motifs (TATA, CCAAT-box, etc).