UCNEbase documentation content
- UCNEbase methodology
- Definition of Ultra-Conserved Non-coding Elements (UCNEs)
- Definition of Ultraconserved Genomic Regulatory Blocks (UGRBs)
- Identification of human UCNE paralogs
- Identification of paralogous UGRBs
- Detection of UCNE orthologs in other species
- Identification of syntenic subclusters of UCNEs in vertebrate genomes
- Identification of possible target genes
- UGRB and UCNE nomenclature
- Database content
- User interfaces
- UCNEbase schema diagram (ER)
- Glossary
UCNEbase methodology | |
|
The information provided by UCNEbase is generated by a combination of automatic procedures and manual curation steps. The methodology used for the creation of UCNEbase is schematically shown in the figure below.
Definition of Ultra-Conserved Non-coding Elements (UCNEs)We defined UCNEs as non-coding human DNA regions that exhibit ≥ 95% sequence identity between human and chicken and are longer than 200bp. The sequence identity threshold corresponds to a base substitution rate of approximately 1% per 100 million years. We have previously shown that sequences fulfilling such stringent criteria exist only in vertebrates. To compile a list of human UCNEs we scanned whole genome alignments between human and chicken downloaded from UCSC genome browser with a sliding window technique. Human and chicken were selected as reference species for two main reasons: (i) their evolutionary distance provides high specificity in detecting functional elements and (ii) both genome assemblies are of high quality and thus suitable for identifying large syntenic regions. From the initially extracted set of ultraconserved sequence elements, we eliminated coding regions, and a few human repetitive sequences aligning with the same chicken sequence. The remaining 4’351 sequences composed our reference set of UCNEs. Each element of this set was then classified as either “intergenic”, “intronic” or “UTR-associated” according to the human gene annotation from RefSeq. The length of the UCNEs identified in this way ranged from 200 - 1419 bp with a mean = 325 bp and a median = 283 bp. The total length is 1.4 Mbp. Definition of Ultraconserved Genomic Regulatory Blocks (UGRBs)We defined UGRBs (also referred to as UCNE clusters) as arrays of UCNEs that are syntenically conserved between the human and chicken genomes. Syntenic conservation means that the orthologs of the individual UCNEs of a human UGRB occur in the same order within a restricted area of a chicken chromosome.
During the initial scan, we required that neighbouring UCNEs must not be separated by more than 0.5 Mb in both human and chicken. However, a few exceptions to this rule were made during subsequent manual curation based on visual inspections of the genomic context. Identification of human UCNE paralogsHuman genomic regions which exhibit significant sequence similarity to a UCNE are considered paralogs of that UCNE. In general, conserved non-coding elements have very fewer paralogs compared to protein-coding genes. However, the relatively rare cases of UCNE paralogs are highly informative with regard to the origin of UGRBs.
To identify human paralogous regions of UCNEs we first split the human genome sequence (hg19) into fragments of 10’000 bp overlapping by 2’000 bp. We then used the program SSEARCH v34 from the FASTA package to compute the optimal local alignment score between each human UCNE and each other human genomic fragment. The following base substitution matrix was used for this purpose:
Identification of paralogous UGRBsThe identification of paralogous genomic regulatory blocks was mostly done by manual curation. As a minimal condition, we required that two UGRBs share at least one paralogous gene. However, most paralogous blocks also share paralogous UCNEs. In some cases, synteny across paralogous blocks was used to redefine the extension of individual UGRBs. In total, 82 UGRBs were found to have at least one paralogous UGRB forming 39 groups. Detection of UCNE orthologs in other speciesCurrently, UCNEbase contains information about UCNE homologs (orthologs and paralogs) in 18 vertebrate genomes:
We also identified a few UCNEs (mainly located in UTRs) that have orthologs in:
To find homologs (orthologs and paralogs) of UCNEs in other species, we used essentially the same protocol as for finding paralogs in the human genome. We split the genomes of a given target species into fragments of 10’000 bp overlapping by 2’000 bp and then used SSEARCH to compute optimal local alignment scores between each human UCNE and each genomic fragment from the target species. For purely technical reasons, we used SSEARCH version v36.3.5 instead of version v34 for this task. We further used SSEARCH instead of PRSS3 to compute base composition-adjusted E-values for each match by shuffling the DNA sequence from the target genome 500 times in windows of 20 bp. All genomic regions matching a UCNE with an E-value ≤ 10-4 were accepted as homologs. To distinguish orthologs from paralogs we proceeded as follows. If the human UCNE had no paralogs, the matching region was immediately classified as an ortholog. Otherwise, the homologous region from the target genome was compared to all human paralogous regions of the UCNE under consideration. Base composition-adjusted E-values were compared as described above. If the sequence matched a paralogous regions with a lower E-value then the UCNE itself, it was considered a paralog. Otherwise, it was considered an ortholog. Identification of syntenic subclusters of UCNEs in vertebrate genomesFor each human UGRB, we identified orthologous syntenic subclusters of UCNEs in other vertebrates. An orthologous syntenic subcluster is a set of UCNE orthologs that occurs as a cluster on the same chromosome, scaffold, or contig in another vertebrate genome assembly such that any two neighbouring UCNEs are separated by ≤ 0.5 Mbp. For most species we would expect only one orthologous cluster per UGRB. In reality, we often find one cluster plus a few isolated orthologous UCNEs located on sequence contigs not assigned to chromosomes. The situation could be different in the five fish species that have undergone a lineage-specific whole genome duplication. Identification of possible target genesGenomic regulatory blocks are generally assumed to control only one target gene belonging to the so-called trans-dev family. With all the information on orthologous and paralogous regions in other genomes at hand, we tried to identify the most likely target gene for each cluster. To this end, we primarily relied on a genomic context analysis approach. We reasoned that target genes will always be conserved together with UCNEs after whole genome duplication events. Based on the analysis of the gene content of paralogous UGRBs in human, and the fate of UGRB-associated UCNEs in duplicated fish genomes, we were often able to identify a single target gene. In the cases where we were left with several candidates, we gave preference to genes encoding transcription factors. In fact, the overwhelming majority of target genes uniquely defined genomic context analysis turned out to be transcription factors, most of them containing either zinc fingers or homeodomains, or both. | |
UGRB and UCNE nomenclature | |
|
In UCNEbase, we try to define names that carry some information about the function and genomic location of UCNEs, as well as its evolutionary relationship to other UCNEs. UCNE names are typically composed of two parts:
UGRBs have the same name as their putative target genes. Elements are identified by common people's names or names from mythology. Within a UGRB, the alphabetical order of the elements reflects the linear arrangement of the elements along chromosome. Importantly, paralogous UCNEs share the same element name (e.g. DACH1_Hana is a paralog of DACH2_Hana). For elements that are not part of a UGRB, the corresponding chromosome name replaces the block name, e.g. chr2_Nemo. The rule that paralogs should have the same name extends to non-clustered UCNEs (e.g. chr10_Sherlock is a paralog of CPEB2_Sherlock). A small number of UCNEs are very close to each other and thus could be part of the same functional entity. To specifically mark such cases, UCNEs that are separated by ≤ 50 bp in both human and chicken are given the same element name, however extended by different serial numbers (e.g. DACH1_Scheherazade_1 and DACH1_Scheherazade_2). | |
Database content | |
|
As UCNEbase is organized along two hierarchical levels, there are two types of entries, UCNEs and UGRBs. About 90% of UCNE entries are related to UGRBs. There is only one entry per UCNE or UGRB containing information for all vertebrate species covered by the resource. Content of a UCNE entryEach UCNE entry has two parts, one providing detailed information relating to the human genome, and a second part providing information on homologous elements in other species. The first part of the UCNE entry contains the following data items:
The second part contains information about homologous regions in other vertebrates. The regions are defined by genomic coordinates, and classified as either orthologs or paralogs. In addition, the sequence identity, E-value and bitscore of the local alignments are stored. A web display of a UCNE entry is shown below:
Content of a UGRB entryEach UGRB entry also has two parts, one providing detailed information relating to the human genome, and a second part providing information on homologous elements and clusters of elements in other species. The main section of a UGRB entry contains the following data items:
The section on sequence conservation contains synteny maps of UGRBs across multiple vertebrate genomes. A complete synteny map for a given species consists of one or several syntenic blocks referred to as “subclusters”. The information associated with a subcluster comprises the genomic coordinates, the number of orthologous UCNEs, and the names of these UCNEs. An example of a web display of a UGRB entry is show below:
| |
User interfaces | |
Data accessUCNEbase provides several query mechanisms to find UCNEs and UGRBs based on different search criteria. All entries can be accessed by their chromosomal location in the human genome or by proximity to particular genes via the web links
"Browse UCNE clusters” and “Browse individual UCNEs”.
Data visualizationUCNEbase relies on the UCSC Genome Browser for data visualization. A large part of the information content is available as custom track files. This has the principle advantage that information from UCNEbase
can be explored together with a great variety of genome annotations from other sources. The UCSC browser also serves as a navigation platform. All data items from UCNEbase that can be displayed in a browser window are back-linked to the corresponding UCNE and UGRB entries. | |
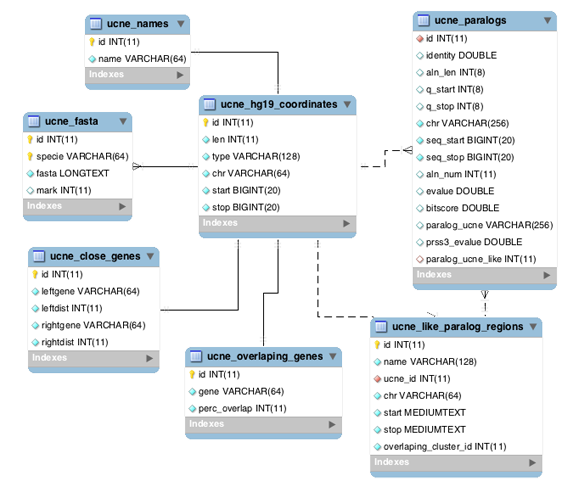
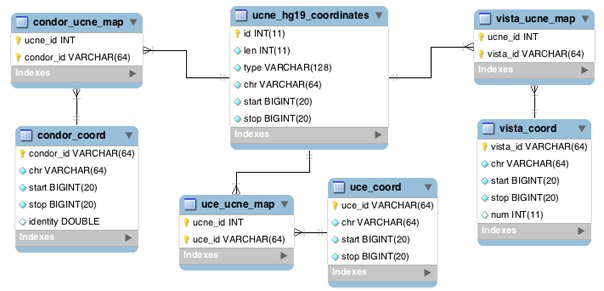
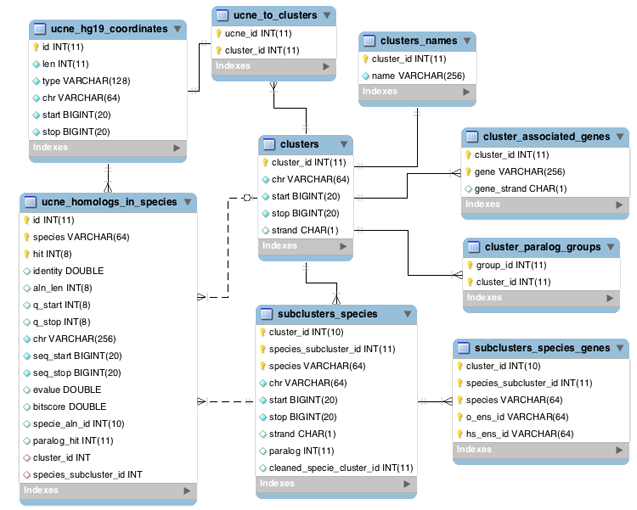
UCNEbase schema diagram (ER) | |
|